
What’s new in biology: spring 2026
A new pancreatic cancer drug, organ recovery advances, weight loss medications protect muscle mass, semaglutide disappoints in Alzheimer's trials, and more.


Niko McCarty and Saloni Dattani review some of the biggest stories in biotechnology and medicine.
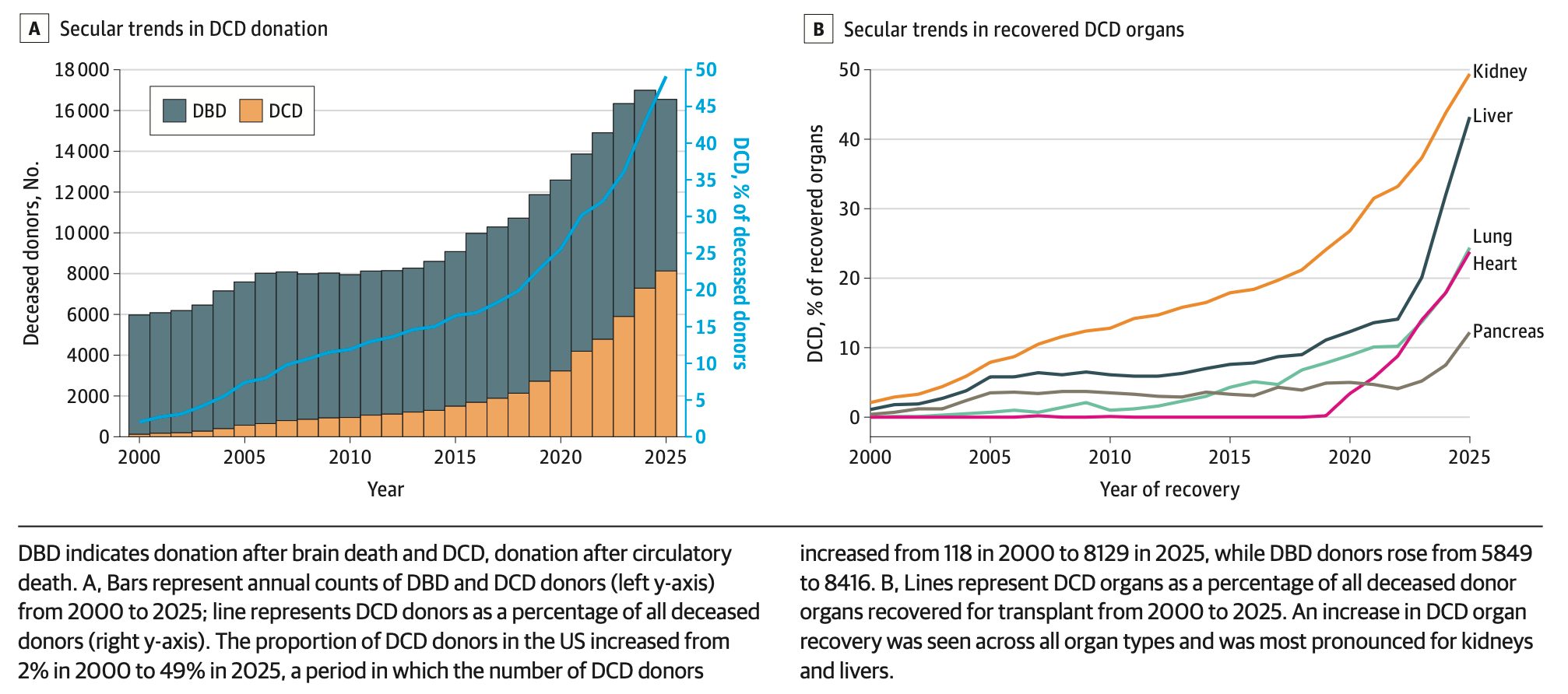
Organ transplants have become much more efficient. Most organ transplants in the US come from deceased donors, and technology has made it much easier to recover those organs and keep them alive for transplants. A new study, looking at all organ transplants from deceased donors, finds a massive rise in the number of organs recovered for donation in the last five years. In 2000, around 2 percent of donors who died from circulatory death were able to donate their organs; that figure rose to 49 percent in 2025.
The authors attribute this to the use of normothermic regional perfusion, a procedure that’s used to restore blood flow to organs after they’re removed from deceased donors, which became more commonly used since 2019, and new machines to perform this procedure for the liver, which were approved in 2021.
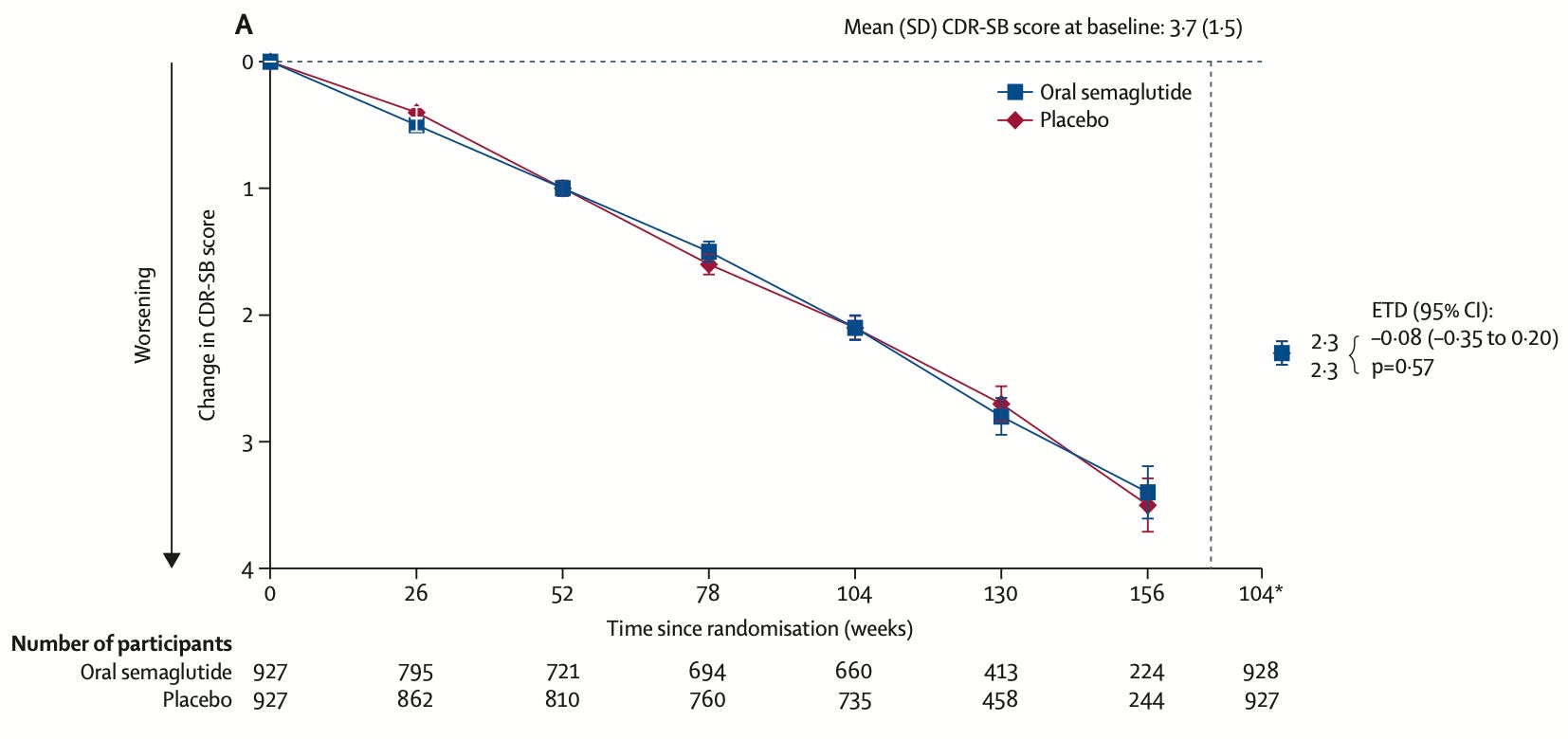
Semaglutide doesn’t slow down Alzheimer’s disease. GLP1 drugs like semaglutide have shown benefits for weight loss, diabetes, kidney disease, and cardiovascular disease, and observational data suggested they might also reduce dementia risk. But two large phase three trials of nearly 4,000 patients with early confirmed Alzheimer’s disease have found no effect on disease progression across cognitive and functional measures.
[Saloni: I expected a slight benefit of GLP-1 drugs through their cardiovascular effects in reducing vascular dementia, which can co-occur with Alzheimer’s, though not a direct benefit.] It’s a useful reminder of the importance of randomized controlled trials, even when there’s plausible evidence or observational data supporting an effect. In this case, it was likely the result of confounding. Sadly, the list of failed Alzheimer’s drugs is long.
A transformative new drug for pancreatic cancer. Pancreatic adenocarcinoma is one of the deadliest cancers, with a five-year survival rate of just 13 percent; advances in therapy against the disease have been slow. A key reason is that around three-quarters of these cancers are driven by mutations in KRAS, a protein that has been considered essentially undruggable because it lacks an obvious binding site for a drug molecule. A new treatment, called daraxonrasib, gets around this by first binding to another protein in the cell, cyclophilin A, forming a complex that can then latch onto and block mutant KRAS, cutting off the signal that drives tumor growth.
In a phase three trial of around 500 patients with metastatic pancreatic cancer who had already progressed while on chemotherapy, the company reported a median overall survival of 13.2 months with daraxonrasib versus 6.7 months with chemotherapy, roughly doubling survival.
That is an unprecedented result for a cancer that has so far been devastating and stubbornly resistant to progress. These are topline numbers released by the company, and detailed data on safety and subgroup analyses will help understand the result better. The drug also appears to cause significant side effects, including serious bleeding and skin problems. But it may also be one of the biggest cancer breakthroughs of the year, if not decade.
This trial enrolled patients with late-stage pancreatic cancer, but phase three trials are also underway testing the drug as a first-line treatment and as an add-on for patients whose tumors are still surgically treatable. The drug is also being tested in lung cancer, and could be expanded further; KRAS mutations drive roughly 20 percent of all human cancers. Since a viable mechanism for targeting KRAS has been demonstrated to work in patients, researchers can focus on iterating, improving safety, optimizing dosing, testing combinations, pushing the drug earlier in the disease course, and expanding its use for other cancers.
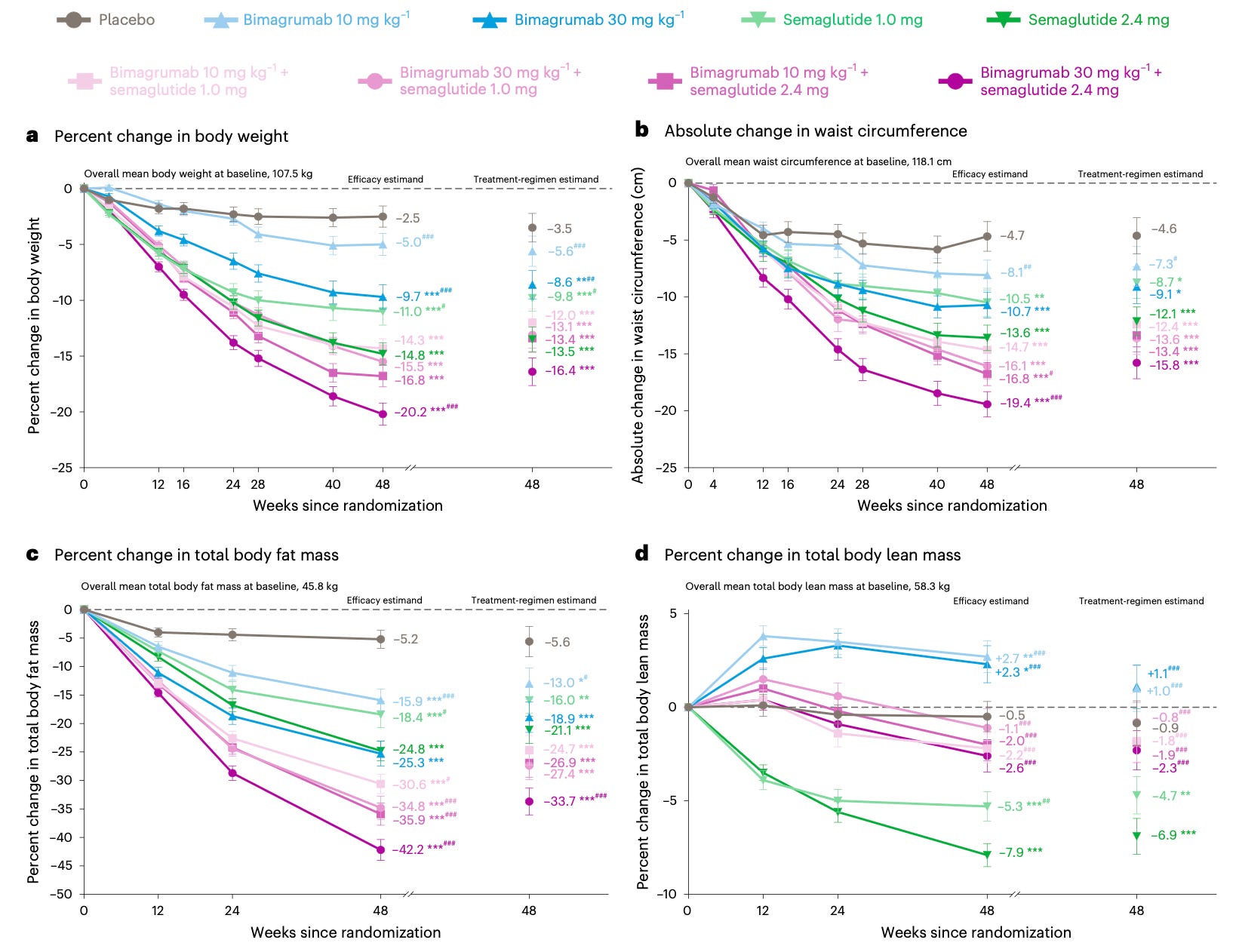
Weight loss drugs that also preserve muscle mass are moving through the pipeline. A well-known side effect of GLP-1 drugs like semaglutide is that around a quarter of the weight lost comes from lean tissue rather than fat, because the drugs work by suppressing appetite rather than directly targeting fat.
A new phase two trial tests whether combining semaglutide with bimagrumab, a new antibody, can address this. Bimagrumab blocks activin receptors, which normally promote fat storage and limit muscle growth, and the idea is that the two drugs work through distinct mechanisms, so their effects on fat loss should be additive, while its muscle-preserving action could offset muscle loss from semaglutide. The trial found that people lost much more fat on the combination treatment than with semaglutide alone, 34 percent compared with a 21 percent reduction in fat mass, while they retained most lean mass, with a 1–2 percent reduction compared to a 7 percent in the semaglutide-only group.
It’s worth noting that bimagrumab has its own drawbacks: it’s given by intravenous injection, which will likely reduce its usage, and likely caused muscle spasms and acne in roughly half the patients in the trial.
Cell simulation caveats. Researchers built a computational model that simulates one cell division, about 100 minutes of biological time, for a single bacterial cell. Each simulation takes four to six days to run on two NVIDIA A100 GPUs and includes some randomness, meaning each simulation plays out slightly differently. After running the simulation 50 times and averaging the results, researchers found they could make a few predictions about how cells work without fitting to experimental data. For example, the simulated cells divided every 105 minutes, which matched experiments, and mRNA molecules had an average half-life of 3.63 minutes, which is close to ground truth.
The cell they’re modeling is JCVI-syn3A, a highly-engineered bacterium with a minimal genome of just 493 genes, compared to over 4,000 for E. coli. Its proteome, transcriptome, and metabolism have all been studied in depth, making it a really good starting point for whole-cell simulation. Rather than simulate every molecule individually (which would vastly exceed the capabilities of any modern computer), the authors instead wrote down all the stuff that happens inside a cell, like transcription, translation, and metabolism, and decided which type of mathematical model would be best-suited to describe each thing. Some cell processes were modeled deterministically, others had ‘spatial’ elements, and other parts were relatively random.
The final simulation had four parts: A Reaction-Diffusion Master Equation (RDME), used to model the individual proteins, RNAs, and ribosomes; a Chemical Master Equation, used to model things where spatial location doesn’t matter as much; Ordinary Differential Equations, used to model changes in metabolite concentrations, and Brownian Dynamics, which simulated the chromosome as a physical chain of beads.
All four models are run together, using a script that synchronizes their results with each other. The Reaction-Diffusion equation takes time steps of 50 microseconds of biological time. Every 12.5 milliseconds of biological time – or after the Reaction-Diffusion equation runs 250 times – the simulation pauses so that the other models can synchronize based on the latest state of the simulation.
This simulation seems to be a major advance over any other existing whole-cell models, but it still does not include a lot of things. For example, the simulation doesn’t account for polysomes, which are clusters of ribosomes that all latch onto a single mRNA and translate at the same time. Polysomes are really common inside of cells, but this simulation assumes that each mRNA can only be translated by one ribosome at a time. Also, it does not include polycistronic transcription. In bacteria, genes are often grouped next to each other on the chromosome and thus transcribed – turned into mRNA – all at once, together. The majority of genes in E. coli, for example, are arranged in these operons, and the authors of this paper acknowledge that many syn3A genes are likely co-transcribed the same way. But the simulation doesn’t capture it.
With that said, this paper is important because by trying to simulate a cell, we are able to figure out what we don’t yet understand and, therefore, which experiments we ought to perform to reconcile those gaps.
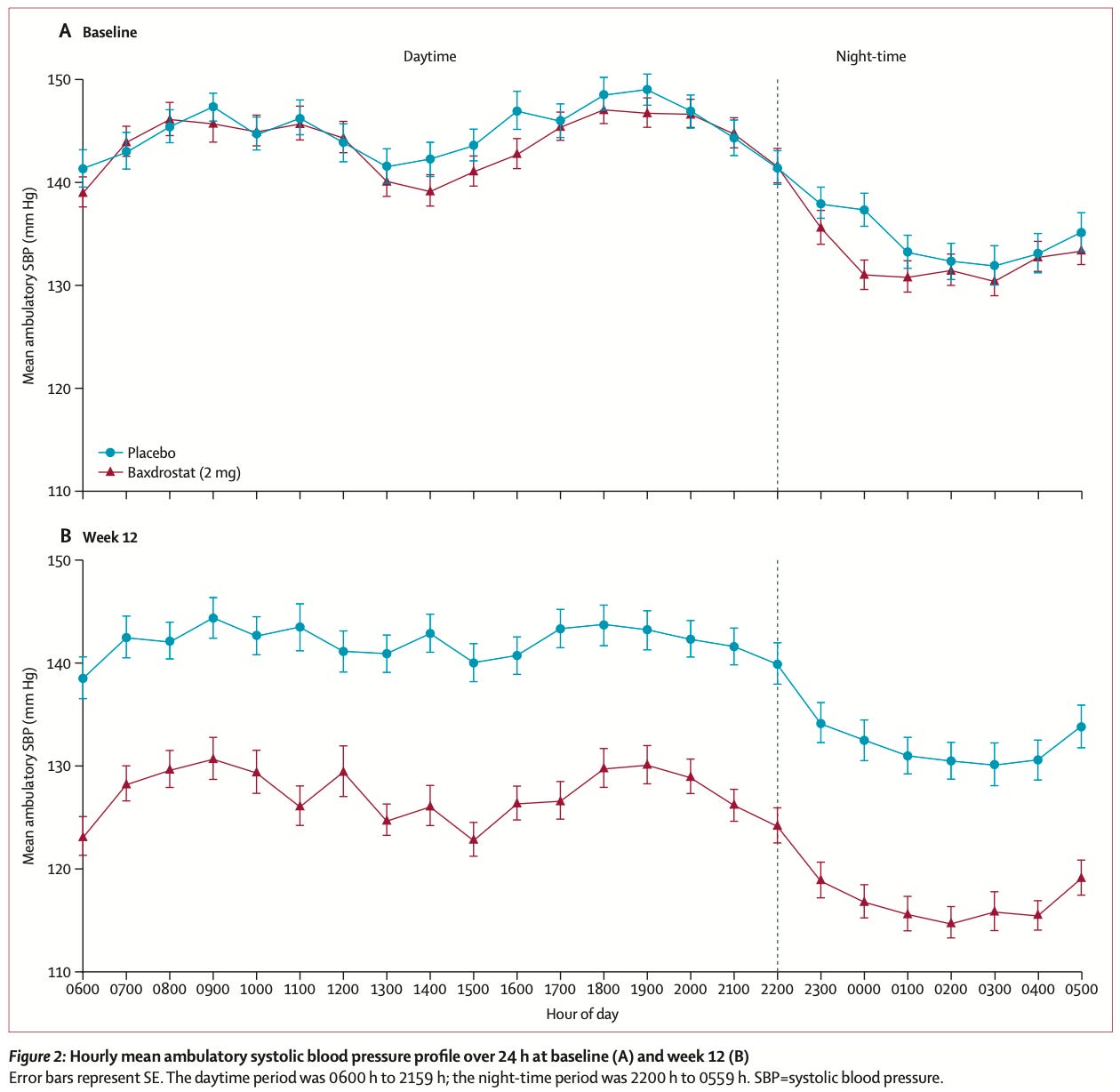
A highly effective new hypertension drug. The body regulates blood pressure partly through aldosterone, a hormone that tells the kidneys to retain salt and water. Some people with hypertension have very high aldosterone levels.
A new drug, baxdrostat, blocks the enzyme that produces aldosterone, reducing how much salt and fluid the kidneys retain. This phase three trial tested the drug in patients with ‘resistant hypertension’, who were already on ≥3 blood pressure drugs but still couldn’t get their readings within recommended levels, <140 mmHg. Researchers measured their ambulatory blood pressure with a monitor worn for 24 hours. That’s much better than single clinic readings, which can be inflated by anxiety and miss overnight blood pressure, which is a strong predictor of heart disease risk.
After 12 weeks, baxdrostat reduced systolic pressure by about 14 mmHg compared to placebo; that makes it one of the largest reductions seen with an antihypertensive drug.
The brain shields tumors from the immune system. For a new study, researchers mapped how a lung tumor talks with the brain in mice. Lung tumors secrete proteins that attract sensory nerves from the vagus, which connects internal organs to the brain. Some of these neurons, which are marked by a specific protein called NPY2R, then send signals from the tumor through the brainstem. In response, the brainstem ramps up sympathetic nerve firing, coaxing noradrenaline to go into the tumor and shield it from attacks by the immune system. When researchers destroyed this circuit, either by silencing the sensory neurons with chemicals or deleting the proteins involved entirely, the lung tumors grew more slowly, the immune system, specifically T cells, was more active, and the mice lived much longer. The next step will be to figure out whether similar circuits exist for other cancers and in other organisms, including humans, before building medicines to disrupt it.
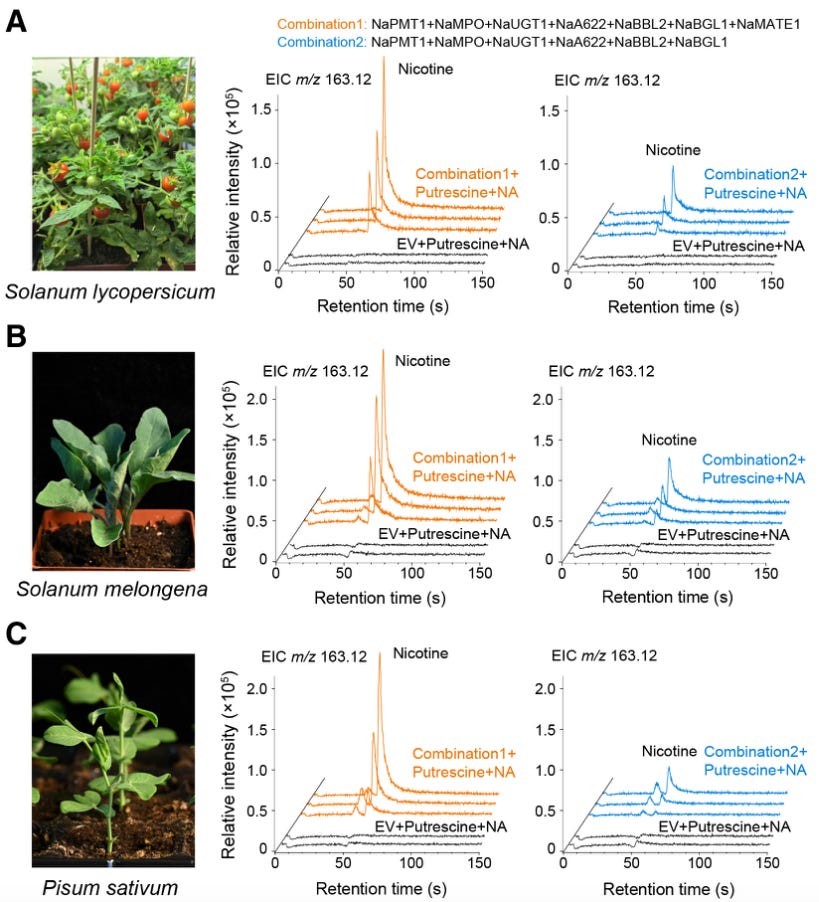
We finally know how plants make nicotine. Nicotine, the molecule, was first isolated nearly 200 years ago by German chemists. It is made from two ring-shaped molecules fused together, but nobody quite knew the enzymes responsible for that final step; the fusion. A new study in Cell, from a group in Shanghai, identifies the enzymes responsible and maps the full pathway for the first time.
An enzyme called A622 strips carbon dioxide from one of the rings, called nicotinic acid, thus making the molecule highly reactive. This reactive intermediate then attacks the other ring, joining the two halves together. Other enzymes strip off a lingering sugar group to make nicotine. Figuring this out took many years of work, as the scientists grew 643 inbred plant lines by crossing together 26 different parent tobacco plants. They finally stumbled upon a mutant plant which was not able to make nicotine, and then sequenced its entire genome. They also crossed back this plant and inbred it for two generations to find the mutation responsible; a single C-to-T swap. With these data and other metabolism experiments (in which plants are fed with heavy isotopes, and those atoms get tracked through the plant), they identified the missing enzymes and biosynthetic steps. And then, just for good measure, the researchers rebuilt the entire pathway in yeast, tomato plants, eggplants, and peas using synthetic genes, just to prove it all worked as they thought.
Did AI cure a dog of cancer? The most viral health story of late concerns Paul Conyngham, a Sydney tech entrepreneur with no biology background who used AI tools to design a personalized mRNA cancer vaccine for his dog, Rosie. Conyngham paid $3,000 to sequence Rosie’s tumor DNA, used AI tools to study the sequence, and then worked with a nearby university to manufacture and inject a custom mRNA vaccine. ‘The tennis ball-sized tumour on Rosie’s hock has shrunk in half’ as a result, according to reporting in The Australian.
Making a one-off mRNA vaccine is not especially hard, as bioengineer Patrick Heizer explains. Researchers cure mice of cancer routinely. It’s much harder to prove that a therapy is both safe and effective in human trials. The $3,000 figure mentioned above is also only the cost of the sequencing itself. The actual cost for these experiments, including reagents and labor, was likely far higher.
Palli Thordarson, Director of the RNA Institute where the work was done, further clarified that not all of Rosie’s tumors responded to the vaccine. The team is investigating whether the dog’s various tumors each mutated in unique ways, which may have made the vaccine less effective against some of them. Thordarson also said the treatment involved the co-administration of a checkpoint inhibitor, anti-PD-1, which is itself a highly effective anticancer drug. This confounds the results, making it unclear whether the mRNA therapy or the anti-PD-1 drug was most useful for Rosie.
This story is most useful, in our eyes, for what it says about the ridiculous, bureaucratic hurdles required for clinical testing, even on dogs. ‘The red tape was actually harder than the vaccine creation, and I was trying to get an Australian ethics approval and run a dog trial on Rosie,’ said Conyngham. ‘It took me three months, putting two hours aside every single night, just typing the 100 page document.’ Similar bureaucracy pervades clinical trials in the US and UK, and we should urgently find faster ways to design and test drugs in human patients.
 | A guest post by
|
Great newsletter!
What an amazing article! I have learned much even though there's much I didn't understand. It was the big picture that awed me. Thank you! Anna