
Pancreatic cancer just met its match
A disease that was once a death sentence is increasingly treatable


For most of the last half-century, a diagnosis of metastatic pancreatic cancer was a death sentence. In December 2025, former Nebraska Senator Ben Sasse announced he had been diagnosed with stage four pancreatic cancer that had spread to his lungs, liver and other organs, and was given three to four months to live from the time of diagnosis. With little to lose, he enrolled in a clinical trial for an experimental drug. Four months later, he reported a 76 percent reduction in tumor volume, describing the drug, daraxonrasib, as a ‘miracle’. His face, ravaged by a severe skin rash from the treatment, told a more complicated story. Yet he was alive and grateful to be able to talk to his family.
A few days after Sasse’s interview, in April 2026, Revolution Medicines announced Phase 3 trial results for daraxonrasib showing the drug had roughly doubled survival in patients with metastatic pancreatic cancer compared to standard chemotherapy. For a disease where median survival has long been measured in months and where little had changed for decades, that result represents a genuine turning point.
But the significance extends beyond pancreatic cancer. Daraxonrasib is among the first drugs in an emerging generation designed to target RAS, a protein implicated in roughly a quarter of all human cancers and long considered beyond reach, in all its mutant forms. And it belongs to a broader class of medicines, molecular glues, that are beginning to show what becomes possible when drugs no longer depend on finding a ready-made pocket in their target. Several compounds in this class are now in clinical development, each probing a different protein that previous generations of drugs could not touch.
Pancreatic cancer: a tough nut to crack
Pancreatic cancer has the highest mortality rate of all major cancers. Although its five-year survival rate has improved from roughly 4 percent in the mid-1990s to around 13 percent today, it remains among the deadliest of all cancer types.
Survival is so poor partially because pancreatic cancer is typically diagnosed late: the pancreas sits deep in the abdomen, symptoms are vague and late to appear, and by the time most patients are diagnosed, the cancer has already spread. This feature has earned pancreatic cancer the name ‘silent killer’. Metastatic cases, where the tumor has already spread to other organs, represent more than half of all new diagnoses. For these patients in particular there has been minimal improvement in outcomes over recent decades, with just 2 to 3 percent still alive five years after their diagnosis.
For decades, no fundamentally new and effective treatments for metastatic cancer emerged. That changed in 2011, when a wave of innovation began transforming the field. At the heart of this renaissance are immunotherapies: drugs that harness the body’s own immune system to fight cancer. Among them are checkpoint inhibitors, which work by releasing the natural brakes on immune activity, and CAR-T therapies, a new class of anti-cancer wonder treatments which engineer a patient’s own immune cells into precision cancer-fighting weapons.
These treatments have redrawn the boundaries of what is possible in oncology. In metastatic melanoma (the most serious type of skin cancer), immunotherapy has produced results once thought unimaginable: from only 25 percent survival after one year twenty years ago to 50 percent survival after 10 years now. Unfortunately, metastatic pancreatic cancer is particularly good at protecting itself against immune attack and has thus remained beyond the reach of this newer wave of drugs.
Pancreatic tumors build a shield around themselves to evade immune attack. They are surrounded by a dense, scar-like layer of tissue that physically blocks a patient’s immune cells from entering the tumor. As a result, checkpoint inhibitors, which work by reactivating immune responses, often have little effect. CAR-T therapies also struggle to penetrate this physical barrier. Even when immune cells do manage to get inside, the tumor creates a hostile environment that weakens them. It does this by attracting suppressive immune cells, including regulatory T cells, myeloid-derived suppressor cells, and tumor-associated macrophages, and by releasing molecules that dampen immune function.
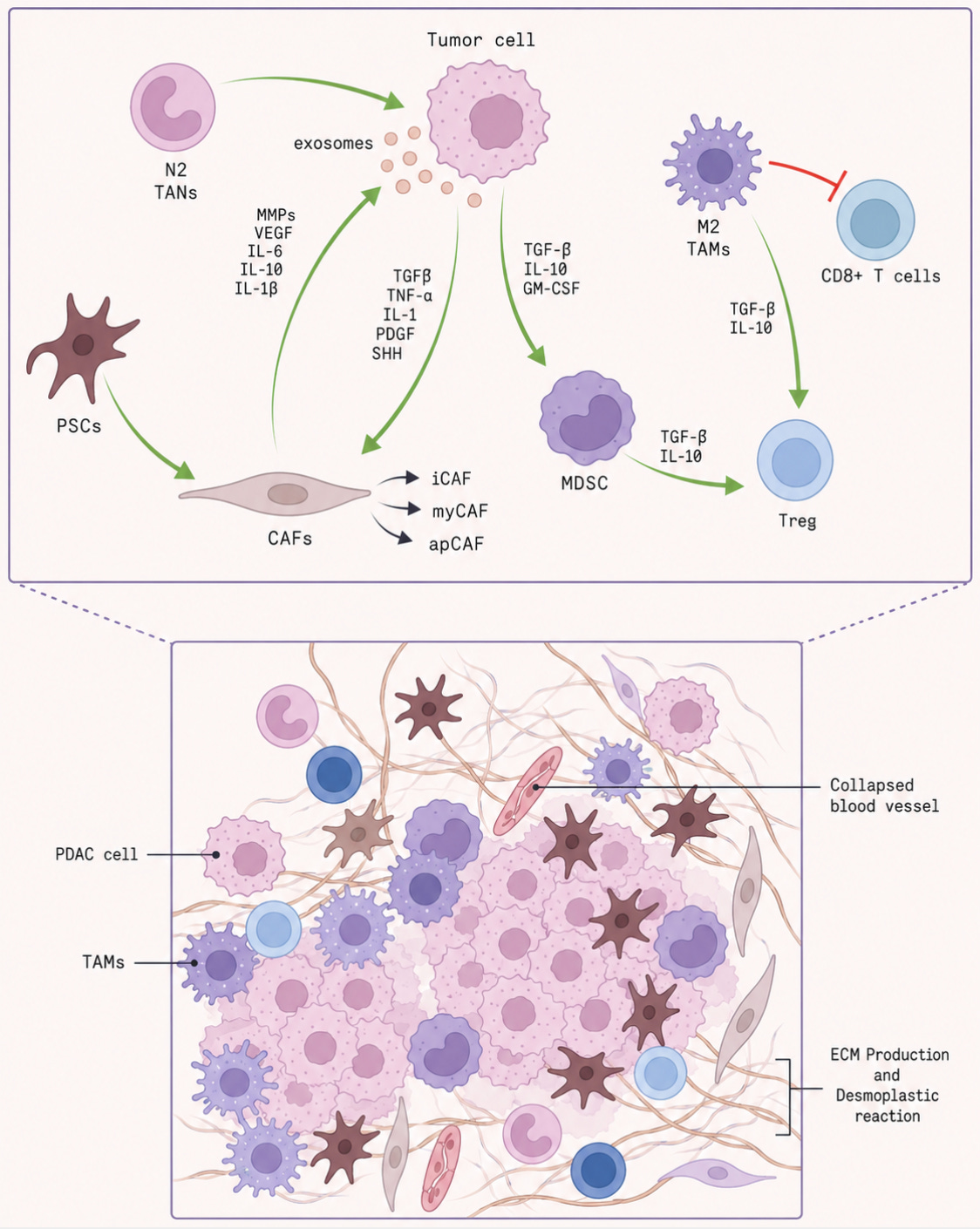
There is a further problem that compounds this. Immunotherapy tends to work best against genetically ‘noisy’ tumors: cancers with many mutations that generate abnormal surface proteins, known as neoantigens, which help the immune system recognise the cell as foreign. Melanoma is a classic example. Driven by UV-induced DNA damage, it often carries a heavy mutational burden and is therefore more susceptible to immune attack. Pancreatic tumors, by contrast, are comparatively quiet. They contain fewer mutations, and can remain largely hidden from immune surveillance.
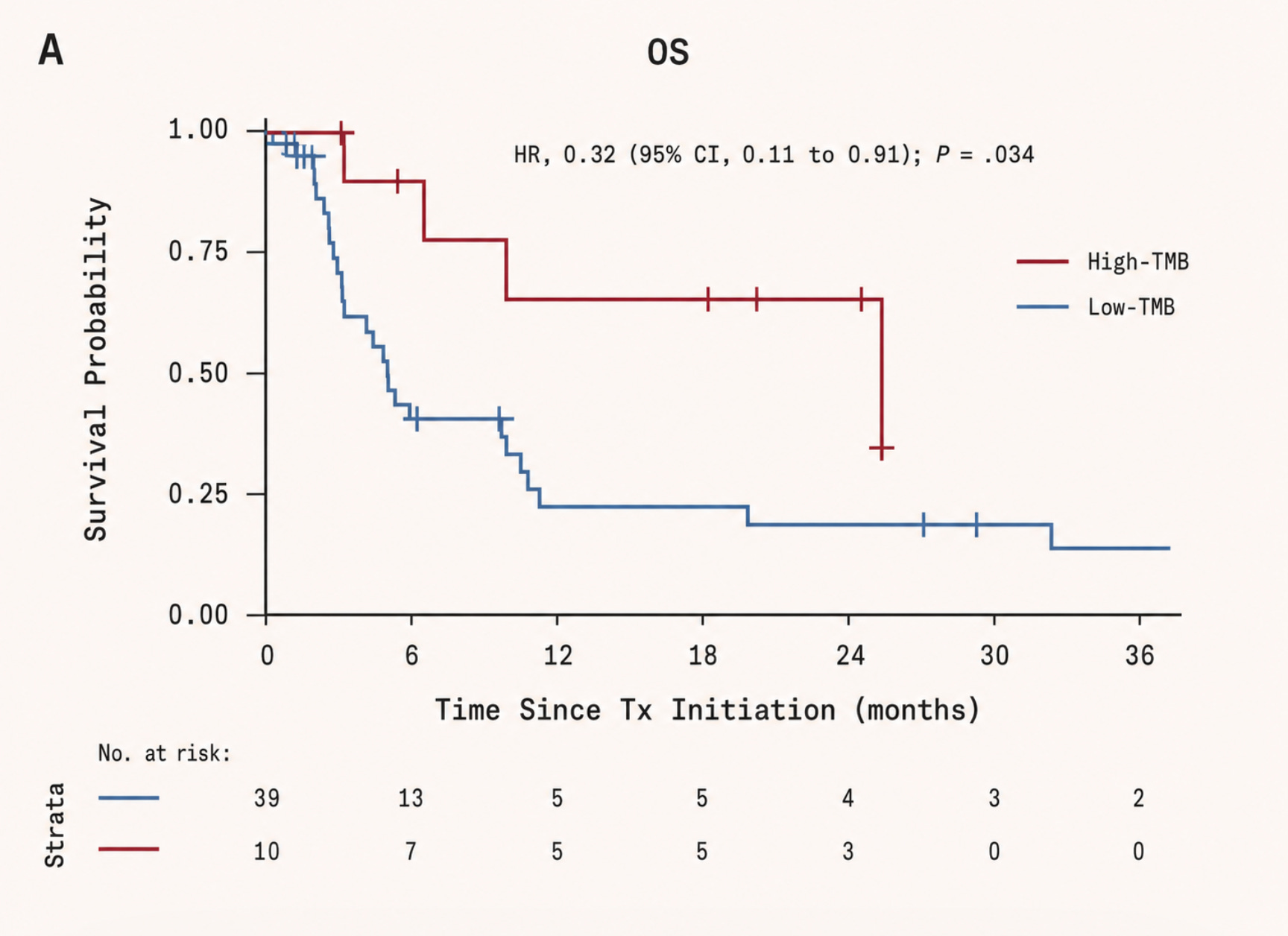
Yet despite these disadvantages, pancreatic cancer has one feature that should, in theory, make it highly targetable: KRAS mutations, which are found in more than 90 percent of cases. A mutation this common, and so central to the cancer’s biology, ought to be a gift to drug developers: a clear and identifiable target around which therapies could be designed. In practice, however, it proved far more difficult.
Targeting RAS
The genomic revolution promised, among other things, that we would crack cancer. When the Human Genome Project concluded in 2003, the optimism was intoxicating. If cancer was fundamentally a disease of mutated DNA, then sequencing that DNA would tell us exactly what to target and how. What researchers hoped is that we would be able to find the broken gene, design a drug to block its protein, and shut the cancer down. Precision medicine, we were told, would transform oncology by swapping the bluntness of the old chemotherapy approaches into something more like a guided missile.
More than twenty years later, the progress has been mediocre. Two problems have stood in the way of realizing the genomic promise. The first is evolution: tumors have genomes that can change rapidly, and under the selective pressure of a targeted drug, resistant clones can emerge and allow the cancer to adapt around the treatment. The second problem is even more fundamental: many of the mutations that drive cancer produce proteins that simply cannot be targeted by conventional drugs.
RAS is one such example. RAS is a molecular switch. In its normal state, it cycles between an active ‘on’ position and an inactive ‘off’ position. When a cell receives a signal to grow or divide, RAS flips on, relays the message downstream through a cascade of other proteins, and then switches itself off again. It is, under normal circumstances, a tightly regulated relay station.
Cancer hijacks this system through mutations, which are small but catastrophic in their effect: they lock RAS permanently in the ‘on’ position. Cells receive a continuous, unrelenting instruction to proliferate, regardless of what the rest of the body is telling them.
For over two decades, RAS carried one of the most dispiriting labels in drug discovery: undruggable. This was not for want of understanding. RAS is in fact one of the most studied proteins in cancer biology, mutated in roughly 25 percent of all human cancers, and in over 90 percent of pancreatic cancers. In lung adenocarcinoma and colorectal cancer, the figure sits between 30 and 45 percent. The problem lies in the fundamental geometry of the protein.
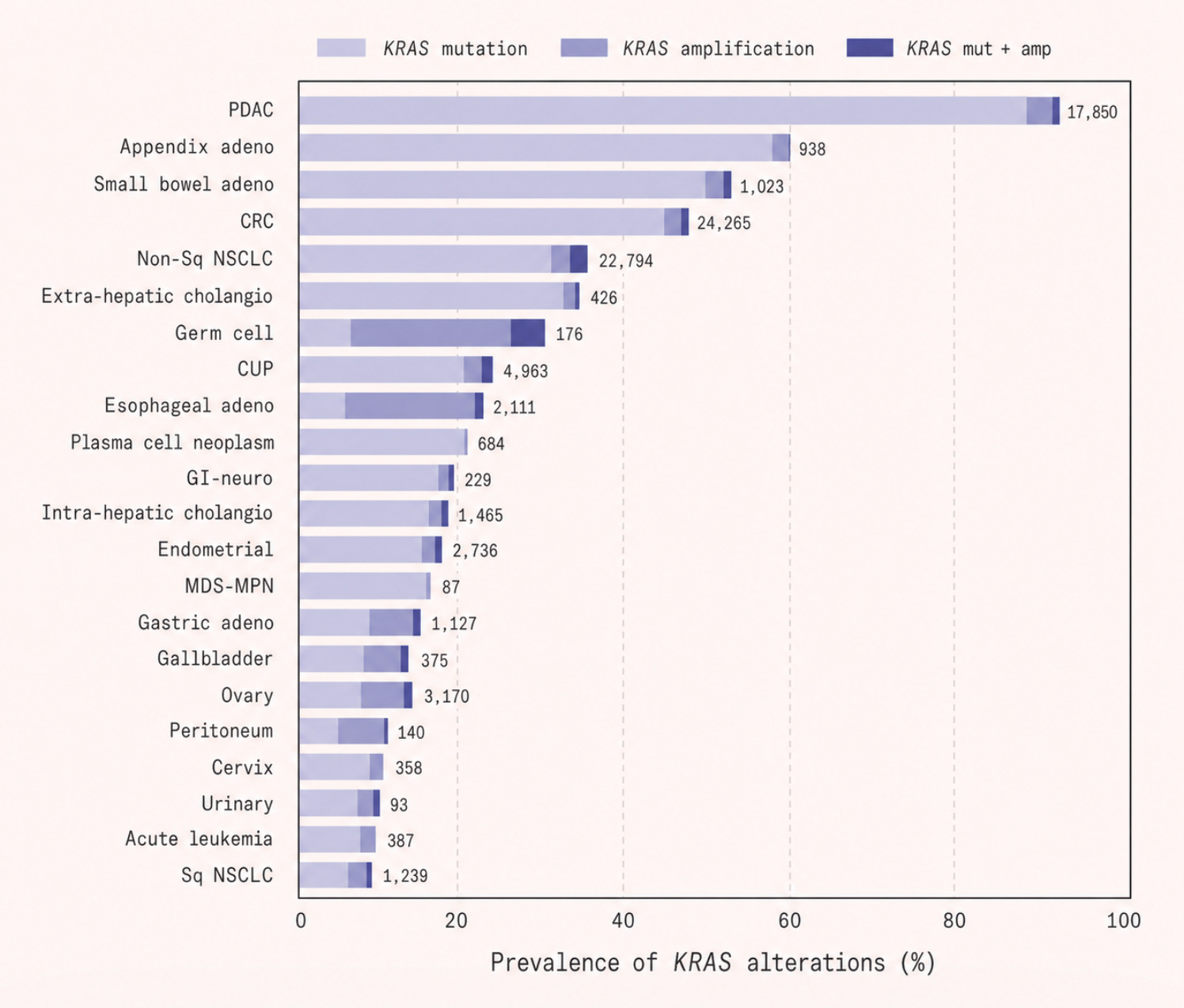
Most cancer drugs are small molecules or compounds precisely shaped to slip into a pocket or groove on a target protein, jamming its function the way a foreign key might get stuck in a lock. This approach depends entirely on the protein offering a suitable cavity to exploit. But RAS’s surface is unusually smooth and chemically inhospitable, presenting none of the clear binding sites that drug designers look for.
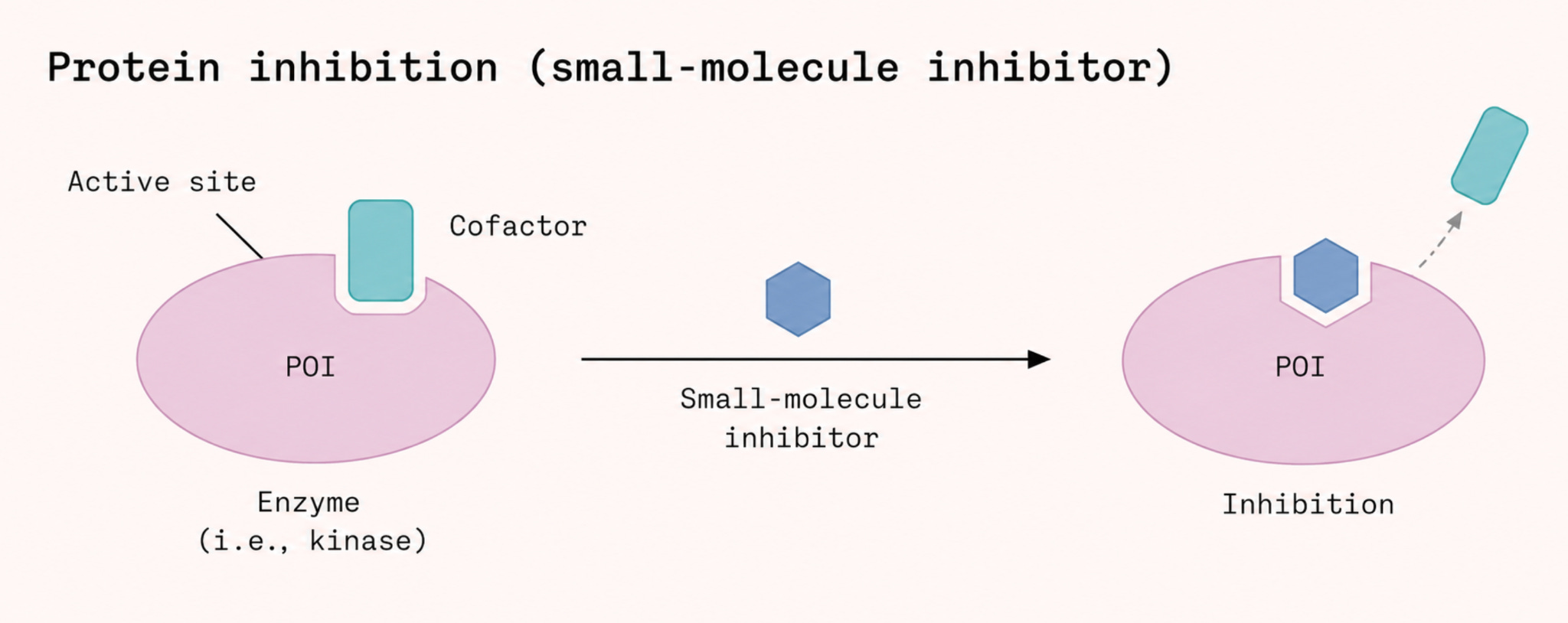
In 2013 researchers demonstrated that small molecules could, in fact, bind to a specific mutant version of RAS called G12C. This particular mutation creates a small pocket that does not exist in the normal protein. The biochemist Kevan Shokat at the University of California, San Francisco, showed that certain compounds could lock into this pocket and trap K-Ras in its inactive state.
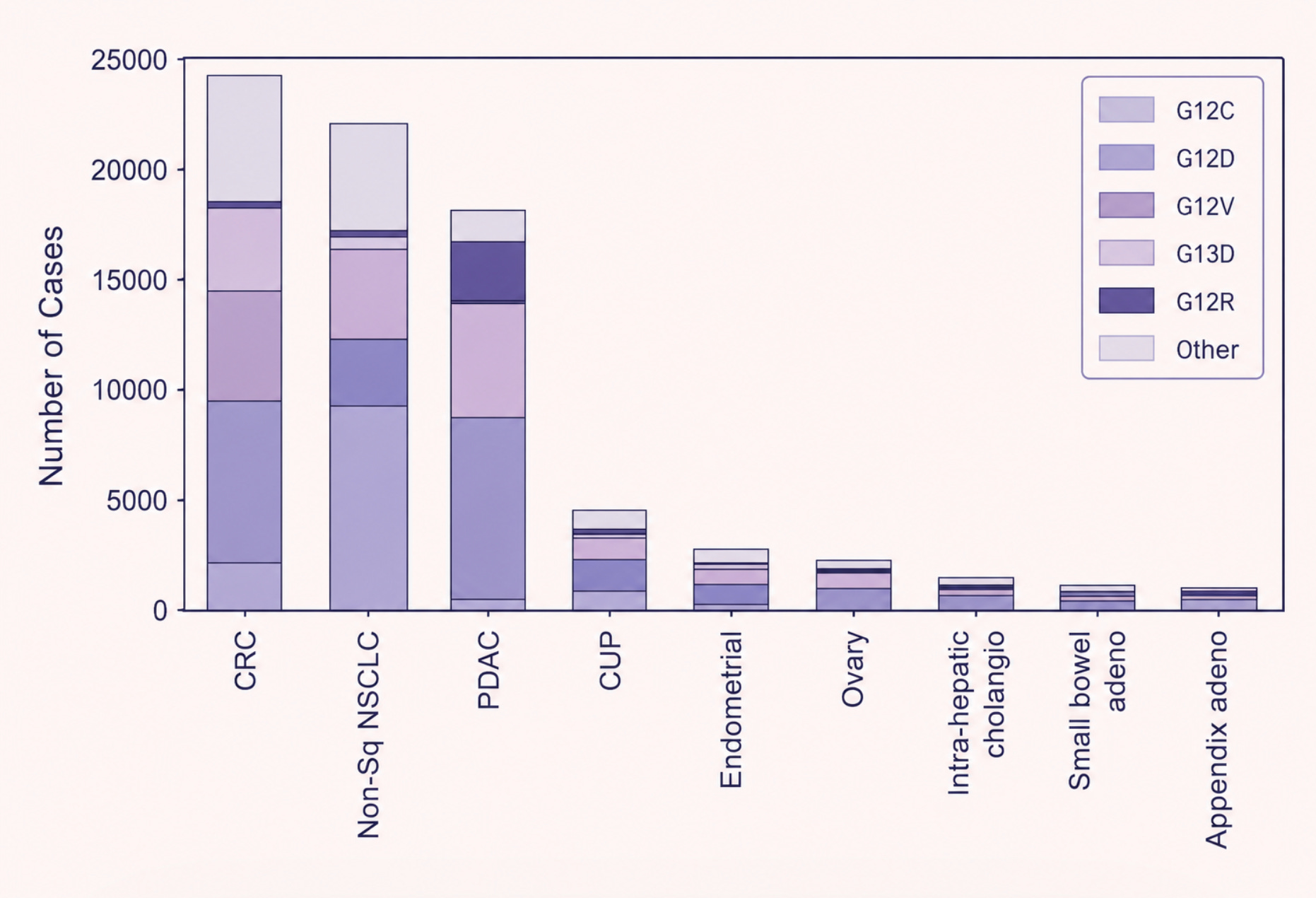
From that insight emerged the first generation of RAS drugs: sotorasib and adagrasib, both of which bind to and ‘switch off’ the G12C-mutated RAS protein. Sotorasib received FDA approval in 2021 for RAS G12C-mutated non-small-cell lung cancer. It was also, for pancreatic cancer patients, largely useless. The G12C mutation is common in lung cancer but rare in pancreatic cancer, accounting for only around 1 percent of cases. The vast majority of pancreatic tumors carry a different RAS mutation entirely, that does not form such a cavity in the protein.
Molecular glue
Molecular glues entered cancer medicine before they had a name. In the late 1990s, thalidomide, a drug infamous for causing birth defects decades earlier, showed unexpected activity against multiple myeloma. Its analogue lenalidomide was approved for multiple myeloma in 2006 and became one of the defining drugs in the disease. Only later, beginning with the identification of cereblon as thalidomide’s target in 2010, did scientists realise that these drugs worked as molecular glues.
Molecular glues take a different approach from most small molecule drugs. Rather than binding a pocket, they create new connections between proteins. A molecular glue binds to the surface of one protein and, in doing so, alters that surface’s chemistry just enough that a second protein is now attracted to it. The two proteins, which might never normally interact, are held together by the glue acting as a chemical intermediary, for example by adding new contact points, or bridging surfaces that would otherwise repel or ignore each other. Because this strategy doesn’t depend on finding a pre-existing cavity, it opens up proteins that have long been considered beyond reach.
After the mechanism of molecular glues was understood, scientists started to think about targeting RAS. But progress required more than understanding molecular glues in principle. Scientists had to recognise that Cyclophilin A, a protein that is common in human cells but with no prior connection to cancer, could serve as a bridging partner, and that a small molecule might hold it against RAS. Seeing whether that was even possible required advances in cryo-electron microscopy, a technique can capture the fleeting, multi-protein assemblies that a molecular glue would need to stabilise. Only once the interface between RAS and Cyclophilin A was mapped precisely enough could a bridging compound be designed.
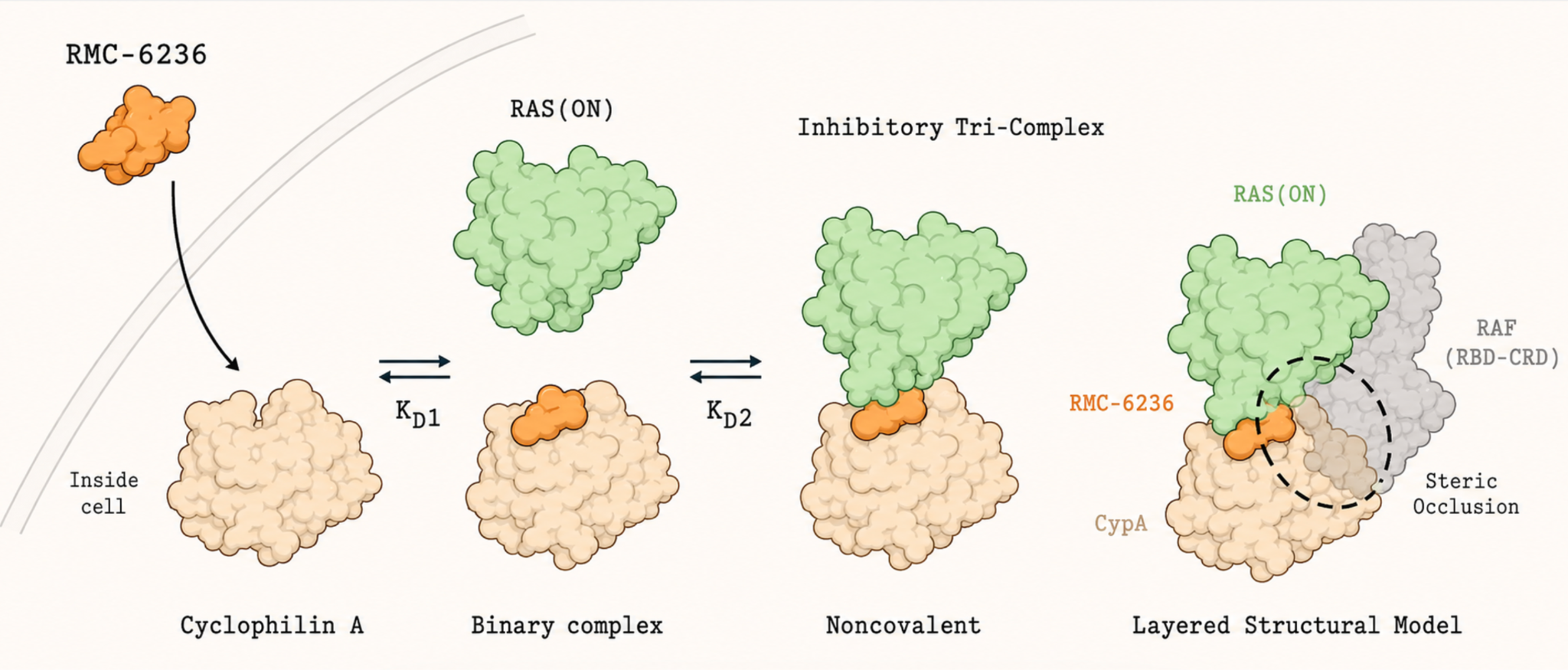
Daraxonrasib is that compound. It binds Cyclophilin A first, and the two form a combined unit whose reshaped surface fits snugly against the active form of RAS, locking the three components together. Cyclophilin A then simply takes up space, blocking RAS from making contact with its downstream partners.
In the Phase 3 trial, patients treated with daraxonrasib experienced a median overall survival of 13.2 months, roughly twice as long as those who received standard chemotherapy: just a prolonging of survival, not a cure. But for a disease that was otherwise so untreatable, doubling survival with a daily pill rather than intravenous chemotherapy is, by the standards of pancreatic cancer, a genuine advance.
The limits of targeted therapy
Daraxonrasib’s clinical success has been real, but resistance inevitably emerges. Analysis of patients treated with the drug has already revealed the tumor’s counter-moves. Some cancers acquire mutations at positions on the RAS protein itself, such as Y64, that physically prevent the molecular glue from forming the complex with Cyclophilin A. Still others amplify copies of KRAS itself, or activate parallel signaling pathways, rerouting growth signals through channels the drug cannot reach.
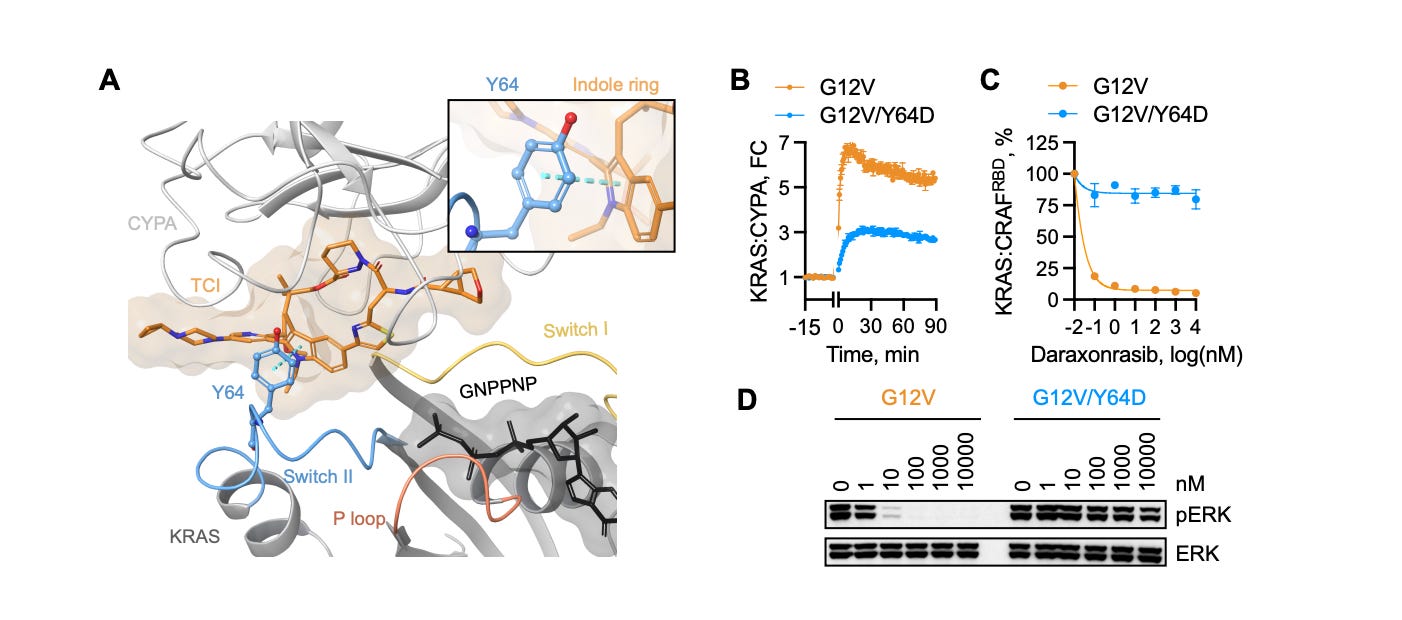
Because resistance to any single agent is essentially guaranteed, the future lies in combinations: hitting multiple vulnerabilities simultaneously, or switching strategies as the tumor evolves. But doing that well requires being able to test new approaches quickly and in small, targeted patient populations. This sits uneasily with a regulatory framework built around several rounds of trials in multiple patients. Matching the right drug to the right mutation in the right patient, in something close to real time, will require that framework to change. One important idea is making small-scale Phase I investigator initiated trials much easier to carry out.
The broader lesson of this era, though, is one of optimism. As well as RAS, the cancer field has scored another win against a previously ‘undruggable’ target, PI3Kα, another commonly mutated protein in cancer. Inavolisib, which both inhibits and destroys the mutant form of the protein, roughly doubled progression-free survival in PIK3CA-mutated advanced breast cancer when added to standard treatment. The assumption that certain targets are simply beyond reach has repeatedly turned out to be wrong.
 | A guest post by
|
My aunt died of pancreatic cancer in January of 2024 after about 18 months with it. I'm sad that another few years might have saved her, but I'm happy that this will happen to a lot fewer families in the future. This is what clinical trial abundance means - fewer deaths like hers.
I am delighted to hear this. My fit 62 year old wife was diagnosed out of the blue with advanced pancreatic cancer in 2009 and died 8 months later, just after our 40th anniversary and the birth of our first grandchild.