
An inconvenient sleuth
Writers such as Nicholas Wade have popularised the idea that Covid-19 originated in a lab, but their arguments are muddled and ignorant of the evidence.

It would be an understatement to say that the pendulum of public discussion has swung in favour of the so-called lab leak hypothesis of the origin of Covid-19. In the latest poll, a majority of the United States public said they believed SARS-CoV-2 originated in a laboratory. Alongside this, ‘fact-checks’ that claimed the virus couldn’t have originated from a lab were quickly archived, retracted, or substantially reframed, while several press articles were edited months after publication without warning.

This scepticism of the ‘official story’ is easy to understand. Scientific and medical authorities made many astonishingly misleading claims during the pandemic, including about its origins. Some of this was politically motivated, with misplaced trust in specific experts, outright falsehoods and now recanted claims. Yet this obviously does not mean that mistakes cannot be made on the other side of the debate, nor does it serve as an excuse to let our guards down about the possibility of overcorrection.
One does not need to look very far to suspect that this has already happened. Going back to public opinion polling, the view that SARS-CoV-2 was engineered in a lab and then released deliberately now outnumbers those who believe there was a purely natural origin, and it is the most common view among those that believe in a laboratory origin.

This is excessive even among popular writers and scientists who believe the virus originated from a lab, however, as it is very rare to find them disagreeing with the idea that the original release was an accident. In contrast, they tend to disagree about whether Covid-19 originated from a laboratory doing general research on viruses, or whether it came from ‘gain of function’ research instead. [1] These positions can be described as the ‘soft lab leak’ and ‘hard lab leak’ positions respectively.
Beyond public opinion, it does matter what the origin of the pandemic is, despite what some may claim. Firstly, there are concerns regarding how much blame should be placed on China. Most people likely believe that an intentional virological research program entails much more ‘playing with fire’, and implies there has been much greater involvement from the government than if the virus originated from a wildlife spillover, due to a government that had not cracked down on unhygienic and unfortunate culinary practices. This should mean that a government should be given much more blame for a pandemic that originates from a lab.
But aside from these obvious consequences, if the pandemic truly originated from laboratory practices involving ‘gain of function’ research, it would also implicate the US NIH, who funded this type of research. This is the claim being made by proponents of the ‘hard lab leak’ hypothesis. Everything from whom we should blame to where we should focus efforts to prevent future pandemics hinges, at least to some extent, on which origins are plausible or demonstrated eventually.
It is unfortunate, then, that this shift in the discussion has come from arguments that are careless or ignorant. Take the now famous piece by Nicholas Wade that argues a lab escape was the most likely origin of the pandemic. It was initially published on Medium before it was republished by The Bulletin of Atomic Scientists, a group concerned with research and technologies that may pose threats to humanity, and famous for their ‘doomsday clock’.
Nicholas Wade is an experienced scientific journalist and, in the piece, he uncovers some conflicts of interest that should have been made known a long time ago. Even if you assign a very low probability to different versions of the lab leak hypothesis, you should agree that Peter Daszak – who was involved in the decision to provide grants for gain of function research on other viruses at the Wuhan Institute of Virology – should not be the main source of claims in this debate due to his conflicts of interests, and perhaps not even a source at all.
Due to these conflicts of interest, Wade dismisses the statement by scientists in the Lancet in February 2020 that argued that China had acted diligently and effectively to share their data with the international community. But once he runs into the letter sent to Nature Medicine in March 2020 by Kristian Andersen et al., arguing that the evidence is in favour of a natural origin of the virus, he has no choice but to engage with the science.
Unfortunately, the quality of Wade’s arguments deteriorates once he shifts to talking about molecular biology, viral evolution, and laboratory practices – making claims that range from outright false to those that are misleading about this virus and the wider issues of evolution and biosafety.
Wade argues that SARS-CoV-2 most likely originated from gain of function research. He explains, firstly, that the most recent methods of viral editing are ‘seamless’, meaning that they would leave no immediately obvious scar sequence, and he argues that if scientists fail to find a similar virus in the wild, this strengthens the case for a lab leak scenario.
This is where the most glaring mistakes and omissions begin. Wade fails to realise how important it is that viral editing still requires a plausible ‘skeleton’ virus to build upon. That is, they require a similar virus to exist in the wild that could be used as the basis of SARS-CoV-2. As a consequence, not finding a close relative in nature harms the case for a lab leak just as much as it harms the case for a zoonotic origin. If we haven’t found similar viruses in the wild, what we can create with current technology stops at step one. While our ability to modify viruses has improved over time, we are nowhere near the level where we can dream up the genome of the virus from scratch, viral capsid and all.
Serial passage
And in trying to find additional supportive evidence to rescue the idea of a lab leak from being pure speculation, Wade fails to convince. Take his claim that the virus might have originated by the ‘serial passage’ of another virus in the lab. This is the idea that a virus has been grown in the lab and gradually cultured in different environments, to cause it to evolve into SARS-CoV-2. If we looked specifically at the sequence of the virus, it is true that artificial selection wouldn’t leave tell-tale signs of this effect – the sequence itself would look natural. But if we zoom out into the wider phylogenetic trees of these viruses, the story changes entirely.
Here’s where it pays to have a refresher on some basic genome biology. The genetic code determines how specific bases in DNA are converted into strings of messenger RNA. Blocks of three bases each, known as codons, get translated into amino acids. Amino acids are the building blocks of proteins behind most of your non-water weight and are responsible for most biological function, whether you are a human or a virus.
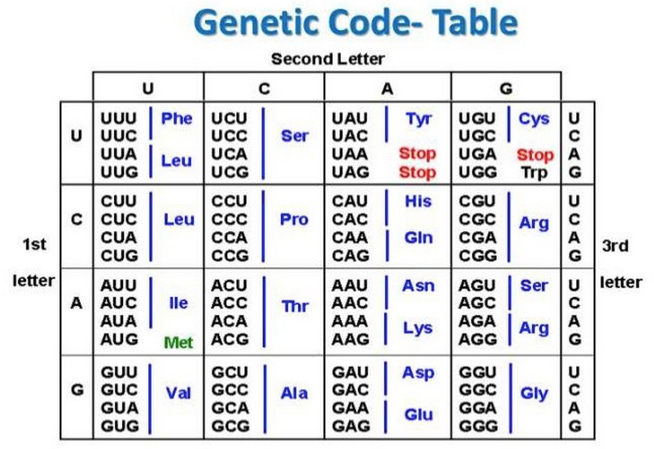
Since there are four bases to choose from (A, C, T and G), there are 64 possible combinations that you can make from a sequence of three DNA bases. You can see above how the genetic code determines which amino acids should be used to build proteins: codons of three DNA bases result in different amino acids. As you might notice though, the genetic code is redundant: there are multiple codons that can give rise to the same amino acid. Despite there being 64 possible combinations, there are only 20 possible amino acids that can be created from them. Many amino acids can be created from one of multiple codons, such as Arginine (Arg), which can be coded by six different codons, but others, such as Methionine (Met) can only result from one combination.
Mutations that would change the bases of the genetic sequence in a way that changes the resulting amino acid into another amino acid are called ‘non-synonymous mutations’. Those that don’t change the resulting amino acid are called ‘synonymous mutations’, and are grouped together in the table.
Why is this relevant? The mutation process is sometimes called ‘random’, which can be a little misleading. A clear way to describe it is that the mutation process is indifferent to selection. That means that natural or artificial selection can cause mutations to become more or less common because of their effect on the survival or replication of the organism. If there is no pressure from selection, mutations will occur in a way that is completely unrelated to whether mutations are synonymous or non-synonymous. We can use a simple mathematical formula to see this happening. This is called the dN/dS ratio, which is the fraction of genetic changes you observe that are non-synonymous divided by those that are synonymous.
And this is where we can come back to phylogenetic trees. The ratios we would expect to see can vary depending on the environment and the organism, but the value is important because it allows us to easily compare selective pressures.
If we have some values to use as a reference, we can use this ratio to predict: 1) whether some mutations have been clearly advantageous since the virus strains diverged from their the last common ancestors, 2) whether the environment has not selected for specific mutations (i.e. it is selectively neutral), 3) whether negative mutations have been purged (i.e. purifying selection) and 4) whether there have been immense pressures from human-directed evolution. As an example, the alpha variant of SARS-CoV-2, which arose in Kent in the UK last year, was suspected to be more concerning than other strains of the virus in part due to this sort of analysis.
Scientists have estimated the dN/dS ratio for coronaviruses and the results are not consistent with the idea that the virus was manipulated in a laboratory under selective passage. We can use the RaTG13 virus lineage as a comparison, which has a ratio of 0.142 (shown below). This shows that the dN/dS ratio in SARS-CoV-2 is perfectly consistent with ordinary purifying selection in the wild.
The result itself is replicated across regions. If anything, more recent analyses show lower values and nowhere in the SARS-CoV-2 viral sequence (particularly, not the spike nor its receptor binding domain) gets close to the high end of values for viruses in the wild (>0.3), let alone ones consistent with an experimental origin.

From a genetic perspective, manipulating a related virus into SARS-CoV-2 through selective passaging would be a dead giveaway with this sort of analysis, even if it was possible, which is itself unclear. It would be similar to a genetic comparison between domesticated cabbage and wildlife plants, or dog breeds and wolves.
The 2012 Fuscher experiments, in which a strain of avian flu was passaged in ferrets to select for pathogenic qualities until it was much more transmissible and dangerous, are perhaps the most infamous example of a similar experiment. But the closest analogue history lies in experiments conducted in 2004, where the murine coronavirus spike protein was altered by persistent cell infection. This gives us a glance of what we would see if Wade’s explanation was true, and the results are striking: when the virus was subjected to selection, the dN/dS ratio rose all the way up to 3.0 in the spike region, [2] which is much higher than what we actually see in SARS-CoV-2.
Phylogenetic analysis is one tool that scientists have at their disposal to uncover the origin of viruses. It has been pivotal numerous times in history, allowing scientists to diagnose when other pathogens have arisen from laboratories or other artificial sources, such as the cattle arbovirus that seemed to re-emerge unscratched after years and is now believed to have arisen from frozen material. Or the 1977 H1N1 influenza strain that seemed more related to flu strains from the 50s than any contemporary relative, which scientists now believe might have emerged from a failed vaccine trial. What it shows is a result that is simply negative for SARS-CoV-2, and that we can rule out the possibility that the virus underwent selective passage in a laboratory.
The furin cleavage site
Notwithstanding this confusion, Wade then turns to one of the properties of the virus that he believes makes it so nefarious: the furin cleavage site, a sequence that allows the virus to enter into human cells with so much more ease that it is arguably a big reason that it became a pandemic at all. The focus on the furin cleavage site is not surprising to long time followers of the lab leak hypothesis.
Yet, once again, evolutionary context does much to weaken Wade’s argument. His claim – that out of all SARS-related beta coronaviruses, SARS-CoV-2 is the only one with a furin cleavage site – is either not as interesting as he makes it sound or is simply false. Within the genera of betacoronavirus, which includes SARS-CoV-2 and other sarbecoviruses, several independent evolutions of furin cleavage sites have been identified. Sarbecoviruses, incidentally, are a group of viruses that are massively undersampled, as might be expected since their name comes from the recent 2003 SARS epidemic.

Perhaps realising that the argument is not as strong he’d like it to be, Wade then goes on to argue it’s not just the furin cleavage sequence that is unusual, but the way it is coded that points to an artificial origin of the virus. In David Baltimore’s (now retracted) words, this evidence is ‘a smoking-gun’ for the lab leak hypothesis. Explaining why this argument is unconvincing requires another level of sophistication in what we have already run through, with a phenomenon known as ‘codon bias’ or ‘codon preference’.
While bearing in mind that the generation of mutations truly is blind to the eventual results, it is true that selection sometimes acts as if synonymous codons were not truly synonymous. This can be because the genome is affected by things such as the availability of nitrogen in the environment, the fundamental biochemistry of packing a long sequence in very little space, the temperature, and the immune system of the host.
This does little to change the earlier argument about dN/dS ratios. But it does mean that the chances you will observe particular amino acids in nature are not the same as you would expect by counting the possibilities in the table above. To maintain our focus in the amino acids under discussion, take Arginine, which can be produced by six different codons. Despite this, an Arginine doesn’t have a 16%, or one in six, chance of being coded by each of the six synonymous codons in coronaviruses. The true odds that Arginine will be coded by CGG in known coronaviruses actually range from 2% (in feline coronaviruses) to 7% (in HKU9, a common cold coronavirus).
This reduced probability of finding an Arginine coded by CGG in nature might seem suspicious, but whatever the reason CGG codons are typically disfavoured in coronaviruses, finding one in SARS-CoV-2 is surely no smoking gun. Feline coronaviruses also have a furin cleavage site and its sequence only differs from the site in SARS-CoV-2 by a single nucleotide (CGG-CGA as opposed to CGG-CGG). This is where the debate runs into falsifiable claims: if further sampling of bat coronaviruses finds one with a furin cleavage site that is identical in this respect, the case for a hard lab leak is weakened even further.
But the greatest refutation to the argument comes from the entire course of the SARS-CoV-2 pandemic itself. If these codons were somehow ‘forced’ into the virus, we would expect the virus to lose them as it evolved over time, and replace them with other codons that are more typical in coronaviruses and code for the same amino acids. Clearly, the virus has had plenty of opportunity to do so, as it has evolved within hundreds of millions of human hosts.
Yet this has not happened: the virus hasn’t even returned to a whole-sequence ‘equilibrium’, where the arginine would be coded by CGG 3% of the time and by other codons >16% of the time. Rather, the CGGs in the furin cleavage site appears to have been under extreme selection pressure to remain constrained. The codon is present in >99.8% of the sequences that have been obtained over the course of the pandemic, which now total over a million. For whatever reason, it’s clear that Covid-19 strongly prefers CGG in that location even while it has been evolving all over the world, outside of laboratories. Arguments that the codon looks ‘forced’, or would only arise if a scientist was intentionally trying to increase how transmissible the virus was in a human host, miss this key point.
Wade also argues that there is something peculiar about the location of the furin cleavage site, because it is in the S1/S2 region of the spike protein – a region which is believed to be important in making the virus highly infectious. While scientists have not yet identified viruses that are identical to SARS-CoV-2 in these special parts of its sequence, they have found close relatives, such as RmYN02. These tell us that, even in nature, the S1/S2 region has been a hotspot for insertions and recombination, during the evolutionary history of coronaviruses. All it would take is one particularly unfortunate sequence to be transferred over from another virus to get the one we now know.
Finally, it is clear that SARS-CoV-2 was originally far from being optimized to infect and harm humans. This is evident from the now-constant emergence of new variants of concern, of which at least four have been consequential, having demonstrably higher rates of transmission or lethality than the original virus that emerged.
Which possibilities remain?
Proponents of the lab leak hypothesis may complain that we have unduly focused on Wade’s argument. Even granting the arguments we made above, they might say that it is too much of a coincidence that the pandemic began so close to laboratories that conducted research on coronaviruses. And without getting into the technical arguments above, it is possible that the origin of the pandemic is instead a more innocent laboratory accident. Perhaps, for example, patient zero was some laboratory assistant collecting bats to conduct research on. And, either out of negligence, or because the safety protocols for that sort of research were woefully inadequate, they got infected and passed on the disease to others.
The problem is, there are many details that make even ‘soft lab leak’ explanations like this less likely than they may seem. The furin cleavage site, for example, is quickly lost in most artificial versions of the virus that are stored in labs – and in SARS-CoV-2 itself – when they are cultured in Vero cells, the most commonly-used cell line for this kind of research. This massively reduces the timeline when a laboratory accident could have occurred. It is also a situation where empirical knowledge could help us update our beliefs on which source was most likely: if we had an inventory of the cell lines that were used in the WIV and other Wuhan labs, we would be able to address concerns about the Institute’s reliability as a source, and figure out whether it was even possible for the virus to originate from laboratory culture.
Once again, it is illustrative to compare with the examples from the past where a human origin could be actually confirmed. The aforementioned 1977 flu virus, which resembled viruses from the 1950s, could also be labelled as one that was probably manipulated because it was temperature sensitive, a property that would only make sense after certain laboratory manipulations. The fact remains that even if the virus emerged from a laboratory accident, it would be easy to tell. Even the ambiguity we see would be unlikely if the virus came from a lab.
And once we’re playing this game of Bayesian tennis, it’s hard to know where to stop. Yes, the origin was in the one city in China that has a virological research lab, out of hundreds that do not. But what proportion of the population are laboratory workers at risk, versus what proportion consume meat at wet markets? What proportion are citizens who live near the wilderness – where serological studies conducted before the pandemic have shown zoonotic spillovers happen all the time? How often do people in these lifestyles get exposed to bats, compared to people in labs with PPE, even if that PPE is modest and insufficient? How do we account for the other reasons why laboratories are where they are – precisely because of the high diversity of bats and bat coronaviruses in the region, and because other epidemics originated not far from that area, which was known for years to be a source of zoonotic risk? With all of these factors in mind, it is difficult to get a clear picture of how likely the soft lab leak scenario really is.
It is only natural for us to want human tragedies to have causes where human agency is ultimately at fault. Acknowledging this as a real bias is, of course, not enough to dismiss the possibility that any form of the lab leak hypothesis is true. But in exaggerating the likelihood, we risk not only another scientific error driven by politics, but also escalating tensions between China and the US and – crucially – undermining our ability to prepare for a potential future pandemic that could become even worse than this one has been.
Anonymous Pangolin is a biologist writing under a pseudonym.